2019-12-13
12月7日-10日,一年一度的美国血液学会(ASH)如期而至,盛会上来自全世界的血液学工作者发表着最新进展。如拜耳公布了其AAV基因疗法在A型血友病的1/2期临床试验中取得积极数据,Sangamo公布针对β地贫的1/2期临床最新数据,这是继CRISPR Therapeutics和Vertex Pharmaceuticals宣布其CTX001在临床1/2期试验对输血依赖性β地中海贫血症(TDT)和严重镰状细胞贫血症(SCD)中的有效治疗数据后的,基因治疗在遗传类血液疾病中又一捷报。同时今年6月,Bluebird bio公司也宣布其基因疗法Zynteglo被欧盟批准上市治疗β地贫。越来越多的科研团队加入基因治疗地贫的研发队伍,既让我们看到基因疗法治愈遗传疾病的潜能,同时也庆幸这类真正“缺医少药”的罕见疾病逐渐受到社会广泛关注。所以,本期我们就与大家一起走进这类不被大众熟悉的疾病—地中海贫血,重点关注其在国内的情况。
遗传性血液病(hereditary blood diseases)是指遗传因素引起的血液系统造血功能紊乱的疾病,主要包括遗传性红细胞系统疾病,遗传性白细胞系统疾病以及遗传性出血疾病几大类。其代表性疾病主要有地中海贫血(Thalassemia,简称地贫),镰刀形红细胞贫血症(Sickle Cell Disease, SCD)血小板无力症(Glanzmann thrombasthenia),血友病(Hemophilia)等[1]。其中地贫和SCD均属于基因突变使血红蛋白缺陷而导致的遗传性红细胞系统疾病,故常可以用于基因疗法根治!如CTX001采用CRISPR/Cas9技术编辑病人自体CD34+造血干细胞后回输病人体内,从而源源不断的产生正常血红蛋白以达到治疗甚至治愈地贫和SCD的效果。
二、地贫和镰状细胞贫血症属于罕见病吗?
1. 地贫和镰状细胞贫血症的发病现状
地贫和镰状细胞贫血症属于罕见病的说法似乎已经被广泛传播,不过这种说法还有待考量。先看一组数据:1925年,国际上Cooley和Lee 首先描述地中海贫血;我国于1940年首次报道广州3名地贫患者,而后陆续发现于北京,浙江等地区,20世纪中期,一次覆盖全国20个省市(自治区)的90万人大规模血红蛋白病调查,基本阐明了中国长江以南是地贫高发区,尤以广西,广东和海南三省(区)为甚[2,3]。
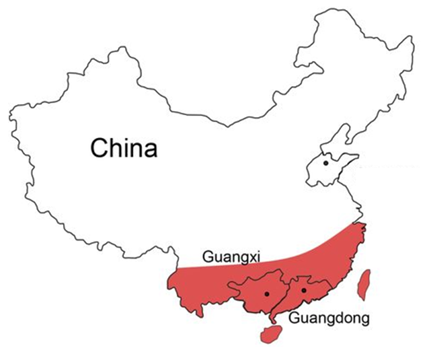
图1:我国地贫主要分布区域[2]
地贫主要分布在包括中国南方在内的全球疟疾高发的热带和亚热带地区,我国长江以南广大地域是地中海贫血的高发区,尤其以广西、广东和海南三省为甚。其中广西地贫发病率最高,人群中每4~5人就有1个地贫缺陷基因携带者,每55个家庭就有1个有重型地贫出生风险,如果没有严格的防控措施,每出生200~250个胎儿就有1个重型地贫患儿;其次是广东,地贫基因携带者大于10%,以2019年人口数1.13亿人计算,地贫基因携带者超过1000万人[3]。
2. 如何定义罕见病?
回到开始的问题上,关于罕见病的定义,是指发病率很低、很少见的疾病,一般为慢性、严重的疾病。但不同国家定义不同:

表1. 不同国家对于罕见病的定义[4]
由上表看到,不同国家对于罕见病定义依国情而不同,但在中国,罕见病一直没有明确的法律释义。2010年5 月,中华医学会医学遗传学分会在上海举办的中国罕见病定义专家研讨会上,与会专家建议将中国的罕见病定义为患病率小于1/500,000或新生儿发病率小于1/10,000 的疾病。据此估算,中国罕见病患者人数约为1,680万。如果按照30万的发病人数计算,地贫刚好大于这个发病率。
3. 地贫可能并不属于罕见病,而镰状细胞贫血症却在列!
2018年5月,国家卫生健康委员会、科学技术部、工业和信息化部、国家药品监督管理局、国家中医药管理局五部门联合公布《第一批罕见病目录》,收录了121种罕见病。这是中国政府首次以疾病目录的形式界定何为罕见病。在这个列表里,我们并没有看到地贫,不过镰状细胞贫血症却在列。因为在我国,镰状细胞贫血症发病率很低,主要集中在黑色人种,如在非洲黑人中的发病率最高,在意大利,希腊等地中海沿岸国家和印度等地,发病人数也不少,目前主要在我国南方发现这类案例。所以地贫是否属于罕见病一直没有明确说法。至少目前数据看来,地贫俨然已成为西南地区的常见病,因此严格防控以及治疗措施的重要性不言而明[5]!
三、地贫的致病机理
地贫最早发现于地中海人群,故称为地中海贫血,又称海洋性贫血或珠蛋白生成障碍性贫血,由于珠蛋白基因表达异常而无法形成正常功能的血红蛋白而引发,是一种常染色体阴性遗传病,是最早在分子水平上阐述其病理学机制的人类遗传病之一。具体致病机理[6]如下:
如下图所示,人体正常血红蛋白结构有2条α-珠蛋白和2条β-珠蛋白分别携带一个血红素后聚合形成血红蛋白(hemoglobin,Hb)四聚体,承担人体内氧运输任务,当α-珠蛋白与β-珠蛋白比例(正常比例为α:β=1)失衡,地贫发病。
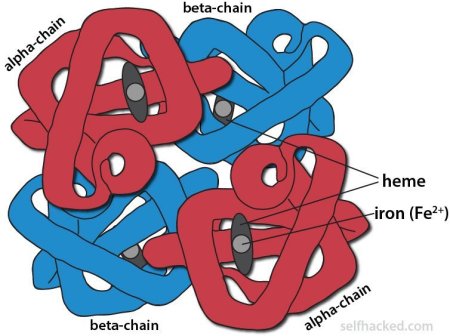
图2. 人类正常珠蛋白基因示意图
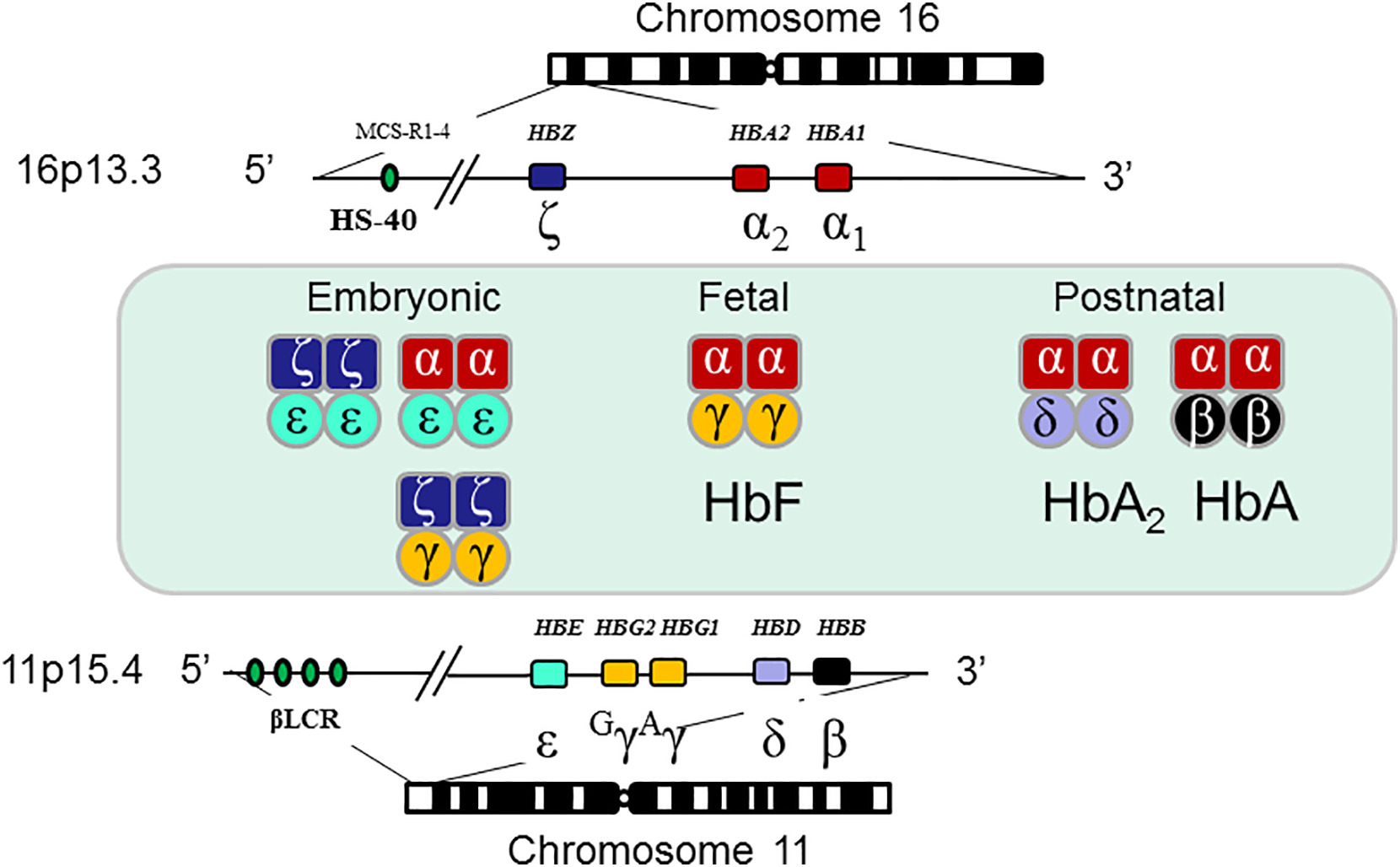
图3. 人类α-和β-珠蛋白基因定位与结构[6]
【注】:α-珠蛋白基因位于16号染色体16p13.3位点,本基因簇含有胚胎期表达基因(ζ)一个,胎儿与成人期表达基因(α1和α2)2个,假基因(ψζ,ψα1)2个,疑似珠蛋白假基因(ψα2,θ)2个,以上α族基因的表达受控于重要调控位点HS-40。同理β-珠蛋白基因簇位于11号染色体11p15.3位点,排列有胚胎期表达基因(ε)1个,胎儿期表达基因( Gγ,αγ)2个,成人期表达基因(β和δ)2个,假基因(ψβ)1个,以上β族基因表达受控于座位调控区(LCR, locus control region)[6]。
1. 地贫的种类
根据异常表达基因的不同,可分为α-地贫,β-地贫,δ-地贫,γ-地贫,δγ-地贫,εγδβ-地贫,其中α-和β-地贫较为常见。此外依据病情轻重不同,β-地贫又分为重型,中型和轻型,α-地贫又分为静止型,轻型,中间型和重型。具体如下表所示:

表2. 根据基因型对地贫进行分类及其表现型[6]
2. 地贫的主要突变形式
α-和β-地贫较为常见,下面主要看这2种地贫的突变形式。α-地贫:主要由基因缺失引起的,如下表(-α3.7/)和(-α4.2/)是中国人最常见的α+-地贫(α-基因功能部分保留记为α+,功能完全丧失记为α0);除了基因缺失,也有少部分是由基因点突变引起的,目前在中国已报道了12种非缺失型α-地贫,其中αWSα/(Hb Westmead,静止型α-地贫)最为常见。
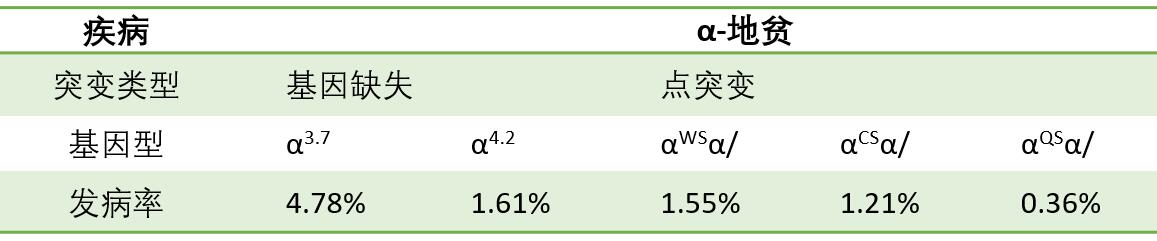
表3. α-地贫主要不同突变形式的占比
β-地贫:主要由基因点突变引起,全世界发现200多种,中国已报道48种。其中常见突变有6种,CD41-42(-TCTT缺失),约占45%,其次是IVS-II654,CD17(A到T点突变),TATA盒子-28等。如下表所示:

表4. β-地贫主要不同突变形式的占比[6]
四、地贫治疗有方?
目前临床上针对地贫的处置方法有输血,铁螯合剂治疗,脾脏切除等等[6](为什么用“处置”而非“治疗”?那是因为以上方法仅仅是起到缓解与维持作用且存在严重副作用或局限性,并不能治愈)。
造血干细胞移植(hematopoietic stemcell transplantation, HSCT):此法是目前唯一能根治重型β-地贫的方法。以人类白细胞抗原(HLA)配型选择供体,方式有骨髓移植,外周血干细胞移植,脐血移植,但因合适的供体来源有限且价格昂贵平均医疗费用40万元左右,临床上难以开展,且影响成功率的因素繁多,不仅与配型有关,还需患者状态即已接受的治疗有关。此外移植并发症如移植物抗宿主病(GVHD),肝静脉阻塞病(hepatic vein occlusive disease,HVOD,简称VOD)、感染、出血和移植失败均增加了此方法的普及难度。
基因治疗--地贫患者的希望!
近日,基因疗法在遗传类血液疾病中频传捷报,让我们看到了基因治疗治愈地贫的希望。目前,针对地中海贫血的基因治疗策略主要分为两种,基于慢病毒整合的方式和基于基因编辑的方式。策略为:递送功能性β-血红蛋白基因拷贝或基因编辑组分至患者的造血干细胞(HSC),从此代替或辅助纠正病变的β-血红蛋白基因完成工作。最新进展[7]如下:CRISPR Therapeutics和Vertex Pharmaceuticals:其资助的编号为NCT03655678的A Safety and Efficacy Study Evaluating CTX001 in Subjects With Transfusion-Dependent Thalassemia项目,在正在进行的1/2期临床试验中取得积极中期数据, 一名输血依赖性β-地贫患者和一名严重镰状细胞贫血症患者在接受CTX001治疗后,均达到停止依赖输血的效果。
这是在美国进行的首个评估CRISPR基因编辑疗法的人体临床试验;Editas公司两项临床前实验项目:于2019年10月7日发布消息,与Cas9对比更加优化的Cas12a-RNP介导编辑CD34+自体干细胞基因编辑获得可喜进展,有望用于SCD治疗。此外于16年发布CRISPR/Cas9用于CD34+自体干细胞基因编辑治疗地中海贫血;MemorialSloan Kettering Cancer Center资助的编号为NCT01639690的ß-ThalassemiaMajor With Autologous CD34+ Hematopoietic Progenitor CellsTransduced With TNS9.3.55 a Lentiviral Vector Encoding the Normal Humanß-Globin Gene项目,基于慢病毒载体TNS9.3.55再编辑CD34+细胞,目前已进入1期试验阶段;
Sangamo Therapeutics资助的编号为NCT03432364的AStudy to Assess the Safety, Tolerability, and Efficacy of ST-400 for Treatment of Transfusion-Dependent Beta-thalassemia (TDT)项目,基于锌指核酸酶(ZFN)技术,再编辑病人自身血液干细胞而后再回输目前已进入2期临床试验。在近日的ASH上也公布了最新数据,表明基因疗法值得进一步探索;Bluebird bio资助的编号为NCT01745120的A Study Evaluating the Safety and Efficacy of the LentiGlobin BB305 Drug Product in β-Thalassemia Major Participants项目,基于LentiGlobin BB305 lentiviral vector编辑病人自身CD34+造血干细胞 已经进入3期临床,且在今年6月,基因治疗药物Zynteglo (LentiGlobin)在欧洲获得批准上市(价格为177万美元),成为继5月份 FDA批准治疗脊髓性肌萎缩的Zolgensma(开发商为诺华,价格约为210万美元)之后第二款超过100万美元价格上市的药物,也是目前全球第二昂贵的药物;
GlaxoSmithKline资助的编号为NCT03275051的Long-term Follow-up of Subjects Treated With OTL-300 for Transfusion Dependent Beta-thalassemia Study (TIGET-BTHAL)项目,基于用编码人β珠蛋白基因的慢病毒载体(globe)遗传修饰的自体造血干细胞/祖细胞分化簇(CD)34+细胞回输病人体内,目前处于临床1/2期试验阶段;IRCCS San Raffaele资助的编号为NCT02453477的Gene Therapy for Transfusion Dependent Beta-thalassemia项目,基于GLOBE lentiviral 编辑病人自体造血干细胞并回输,目前处于临床1/2期试验阶段。关于地贫的介绍本期就到这里了,大家是否对这个疾病又有了重新的认识呢。面对患者群体不断上升,希望我们可以不断推动地贫及其他罕见病在社会上的关注度,让更多患者获得治疗,重返正常生活。
参考文献
【1】李慧媛,杨仁池.遗传性血液病研究进展[J].中国科学:生命科 学,2017,47(12):1306-1312.
【2】叶向化,我国的地中海贫血症[J].铁道医学,1980(09).
【3】中国地中海蓝皮书(2015)
【4】中国罕见病药物可及性报告,2019
【5】中国第一批罕见病目录,2018
【6】《地中海贫血预防控制操作指南》,主编:徐湘民

